Overcoming genome sequencing gaps to accurately characterise a genetically modified mosquito strain

Genome sequencing of malaria vectors has given scientists a roadmap to identify the location of a potential transgene insertion site. However, doing so accurately is compromised by repetitive, unannotated, and fragmented elements on vectors’ genomes. For example, it is estimated that 33% of the Anopheles gambiae genome is composed of repetitive elements.
In a recent study, published in Pathogens and Global Health, my colleagues from Imperial College London, Polo GGB, King’s College London, and I have used multiple methods to extensively characterize and identify the transgene insertion sites in the non gene drive genetically modified male bias mosquito strain. This strain is an intermediate step towards the development of gene drive mosquito as a tool to fight malaria and the molecular characterization of the transgene will support the prediction of dynamics of this modification in potential target field populations.
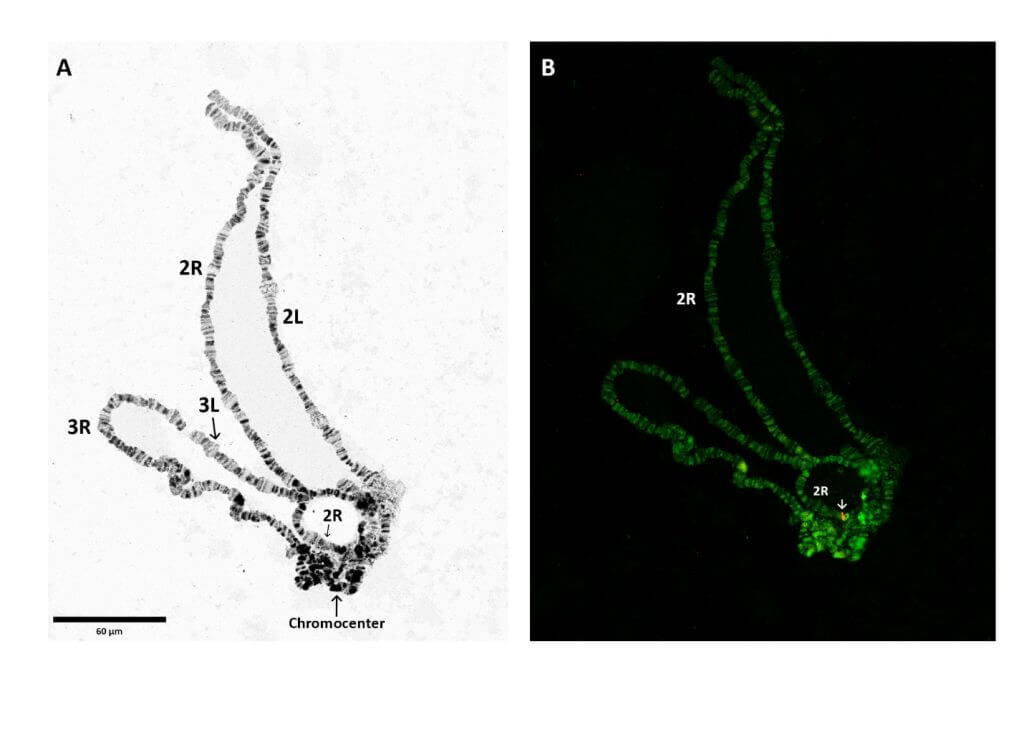
In this study, we combined novel techniques in sequencing and scaffolding1: Whole Genome Sequencing (WGS), Southern blotting, Fluorescence in situ hybridization (FISH) and Polymerase Chain Reaction (PCR) analysis to identify a single insertion of the transgene with the insertion on a single chromosome.
The combination of these methods revealed that the modification was on a different chromosome than was previously described in Galizi, 2014. Based on our findings we concluded that inverse PCR, which produces relatively short flanking sequences, may be less suitable for highly repetitive long DNA sequences (>700 bp) to ascertain correct identification of transgene landing sites. Further, FISH analysis, which does not rely on DNA sequences, can be a very powerful tool to narrow down the chromosomal locations of transgenes in unannotated regions.
These findings were important and showed that combining various sequencing and non-sequencing techniques in this way could support others in the development of novel targets for vector control, off-target analysis, and characterization of insertion sites of genetically modified strains.
The WHO has emphasized in its’ Guidance Framework for testing of genetically modified mosquitoes that for any genetically modified organism that is considered for release, a molecular characterization of the transgene and its neighbouring DNA regions are essential for their safety assessment and post-release monitoring. This study reveals a robust method pipeline that could support this effort.
1 Scaffolding is a technique used in bioinformatics. It is defined as follows: Link together a non-contiguous series of genomic sequences into a scaffold, consisting of sequences separated by gaps of known length. The sequences that are linked are typically contiguous sequences corresponding to read overlaps EDAM – Bioscientific data analysis ontology – Scaffolding – Classes | NCBO BioPortal (bioontology.org)