Combler les lacunes du séquençage génomique pour caractériser précisément une souche de moustique génétiquement modifié

Le séquençage du génome des vecteurs du paludisme a permis aux scientifiques d’établir une feuille de route pour identifier l’emplacement des sites potentiels d’insertion de transgènes. Cependant, la précision de cette procédure est compromise par des séquences répétées, non annotées et fragmentées dans le génome des vecteurs. Par exemple, on estime que 33 % du génome d’Anopheles gambiae est composé de séquences répétées.
Dans une étude récente, publiée dans Pathogens and Global Health, mes collègues de l’Imperial College London, du Polo GGB, du King’ s College London, et moi-même avons utilisé plusieurs méthodes pour caractériser et identifier en détail les sites d’insertion de transgènes dans la souche de moustiques mâles biaisés génétiquement modifiés sans impulsion génétique. Cette souche constitue une étape intermédiaire vers le développement d’un moustique avec impulsion génétique en tant qu’outil de lutte contre le paludisme et la caractérisation moléculaire du transgène soutiendra la prédiction de la dynamique de cette modification dans les populations cibles potentielles sur le terrain.
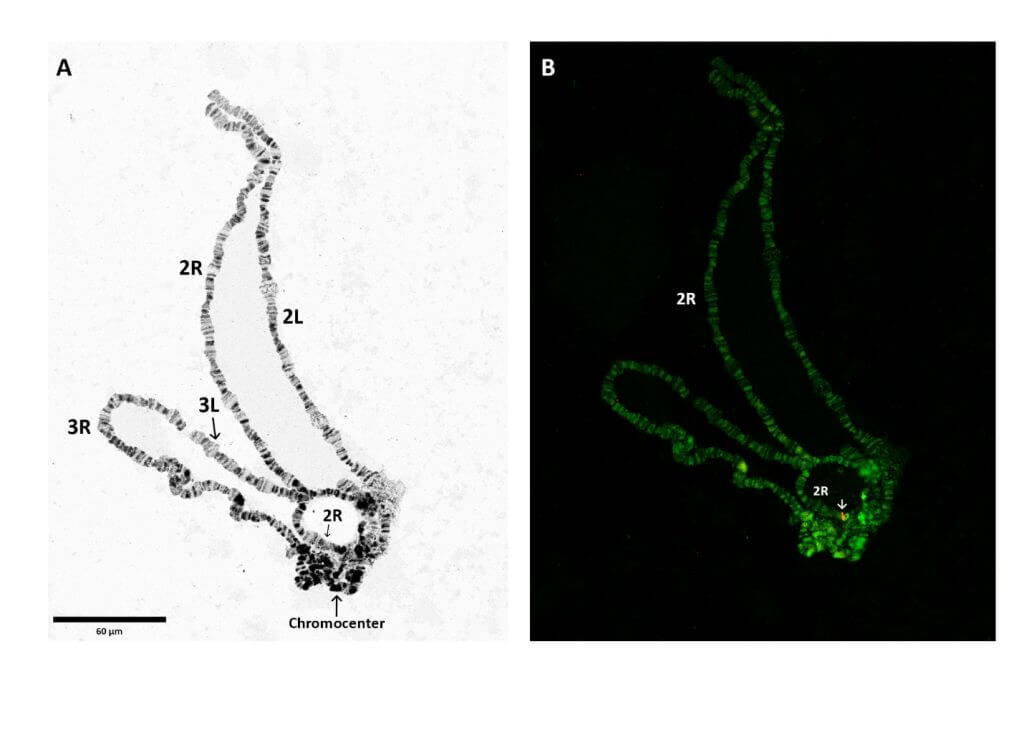
Dans cette étude, nous avons combiné des techniques novatrices de séquençage et d’échafaudage1 : analyse par séquençage du génome entier (WGS), Southern blot (buvardage de Southern), hybridation in situ par fluorescence (FISH) et réaction de polymérase en chaîne (PCR) pour identifier une insertion unique du transgène avec insertion sur un seul chromosome.
La combinaison de ces méthodes a révélé que la modification se trouvait sur un chromosome différent de celui décrit précédemment dans Galizi, 2014. Ces résultats nous ont permis de conclure que la PCR inverse, qui produit des séquences d’accompagnement relativement courtes, est sans doute moins appropriée pour des séquences d’ADN longues hautement répétitives (>700 pb) pour vérifier l’identification correcte des sites d’implantation des transgènes. En outre, l’analyse FISH, qui ne repose pas sur des séquences d’ADN, peut être un outil très puissant pour identifier plus précisément la localisation chromosomique des transgènes dans les régions non annotées.
Ces résultats sont importants et montrent que la combinaison de diverses techniques de séquençage et de non-séquençage peut aider d’autres chercheurs à développer de nouvelles cibles pour la lutte antivectorielle, l’analyse hors cible et la caractérisation des sites d’insertion de souches génétiquement modifiées.
L’OMS a souligné dans son « Cadre d’orientation pour l’évaluation des moustiques génétiquement modifiés » que, pour tout organisme génétiquement modifié dont la dissémination est envisagée, une caractérisation moléculaire du transgène et de ses régions d’ADN voisines est essentielle à l’évaluation de son innocuité et à sa surveillance après la dissémination. Cette étude révèle un ensemble de méthodes concrètes qui pourraient soutenir cet effort.
1 L’échafaudage est une technique utilisée en bio-informatique. Elle est définie comme suit : lier ensemble une série non contiguë de séquences génomiques pour former un échafaudage, constitué de séquences séparées par des intervalles de longueur connue. Les séquences qui sont liées sont typiquement des séquences contiguës correspondant à des chevauchements de lecture.